
© Angelika Kramer
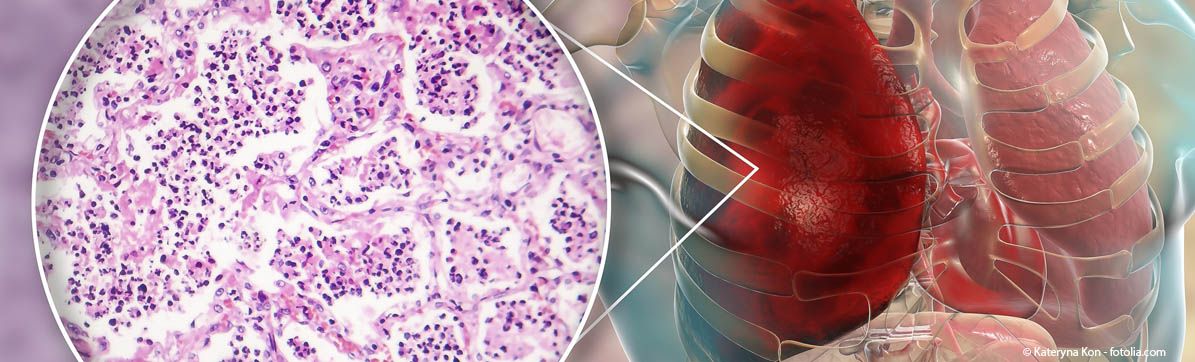
Das Wissen um die Ursachen und die Entwicklung bei interstitiellen Lungenerkrankungen (ILD) (vereinfacht: Lungenfibrose) ist noch nicht vollständig geklärt. Sie gelten als überaus heterogenes Krankheitsbild mit lediglich einer Gemeinsamkeit: Es kommt zu einer vermehrten Bindegewebsproduktion - das Lungengerüst vernarbt, die Lungenfunktion der Patienten verschlechtert sich. Einige Formen der ILD sind unheilbar und enden oft tödlich. Bei manchen ILD-Erkrankungen kennt man die Auslöser: Medikamente etwa, rheumatische Prozesse oder Asbest. Bei anderen ist die Ursache nicht fassbar, sie werden deswegen „Idiopathische Lungenfibrose“ genannt.
Wie sich die Krankheit entwickelt, ist nur schwer zu prognostizieren. Letzte mögliche Therapie ist eine Lungentransplantation – bei dafür geeigneten Patienten. Im Schnitt versterben die Patienten drei bis vier Jahre nach der Diagnosestellung.
Viel Ansporn also für die Lungenforschung, viele offene Fragen, die unsere Forschenden beantworten wollen, um das Überleben der Patienten zu sichern:
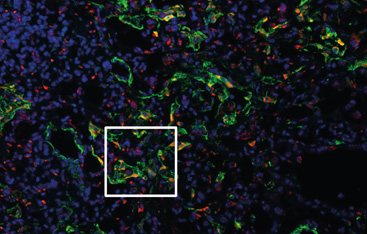
- Generell wollen die Forschenden verstehen, was genau bei der Vernarbung des Lungengerüsts auf zellulärer Ebene passiert. Wo könnte man ansetzen, um diese Vorgänge zu stoppen oder gar rückgängig zu machen?
- In mehreren Klinischen Studien versuchen Forschende des Deutschen Zentrums für Lungenforschung zu ergründen, wie sie IPF-Patienten helfen können: Welche Therapien, welche Medikamente können das Fortschreiten der Krankheit wenigstens verlangsamen?
- Für die seltene Children’s interstitial lung disease suchen Wissenschaftler:innen systematisch nach neuen Therapien. Bei betroffenen Kindern ist die Atmung erschwert, weil das Gewebe rund um die Lungenbläschen geschädigt ist.
- Welche Rolle spielen Autoantikörper bei der Entstehung bei Fibrose? Möglicherweise eignen sie sich als Biomarker um den Krankheitsverlauf vorherzusagen.