
© Angelika Kramer
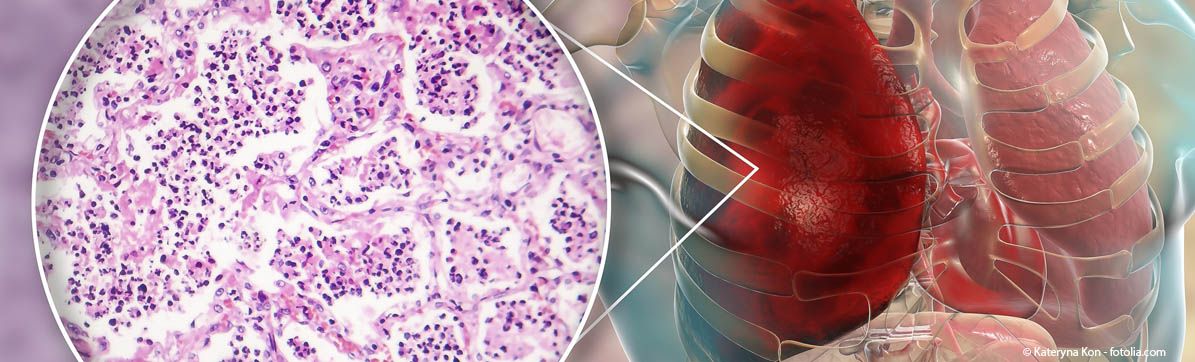
The knowledge about the causes and development of interstitial lung diseases (ILD) (simplified: pulmonary fibrosis) is not yet fully understood. They are considered to be an extremely heterogeneous clinical picture with only one thing in common: there is an increased production of connective tissue - the lung structure becomes scarred and the patient's lung function deteriorates. Some forms of ILD are incurable and often fatal.
The triggers for some ILD diseases are known: medications, for example, rheumatic processes or asbestos. In others, the cause is unclear, which is why they are called “idiopathic pulmonary fibrosis”. How the disease will develop is difficult to predict. The last possible treatment is a lung transplant – in suitable patients. On average, patients die three to four years after diagnosis.
So there is a lot of motivation for lung research, many open questions that our researchers want to answer in order to ensure the survival of patients:
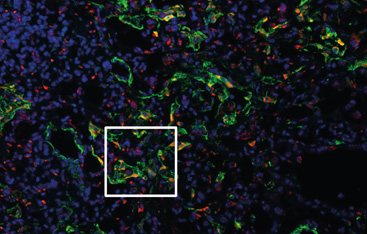
- In general, the researchers want to understand what exactly happens when the lung structure is scarred at the cellular level. Where could we start to stop or even reverse these processes?
- In several clinical studies, researchers at the German Center for Lung Research are trying to find out how they can help IPF patients: Which therapies and which medications can at least slow down the progression of the disease?
- Scientists are systematically looking for new therapies for the rare children’s interstitial lung disease. In affected children, breathing is difficult because the tissue around the alveoli is damaged.
- What role do autoantibodies play in the development of fibrosis? They may be suitable as biomarkers to predict the course of the disease.